Vad är Marfan syndrom?
Marfan syndrom beskriver en komplicerad ärftlig störning i bindväven, som huvudsakligen påverkar ögonen, hjärt-kärlsystemet och skelettmuskelsystemet. Men med tanke på att varje organ består av bindväv, kan Marfan syndrom idealiskt förstöra och störa kraftigt med tillväxt och funktion hos varje anatomisk plats.
Syndromet överförs som ett autosomalt dominerande drag: vi står därför inför en allvarlig genetisk sjukdom, som har ett extremt variabelt fenotypiskt uttryck (defekter kan skilja sig enormt från familj till familj eller från patient till patient).
Det som utlöser Marfan syndrom är förändringen av FBN1-genen (på kromosom 15), som kodar för fibrillin-1, ett mycket viktigt bindande glykoprotein som utgör det strukturella stödet för mikrofibriller.
Mikrofibriller: bestående av fibrillin, mikrofibrillerna är närvarande i den extracellulära matrisen, i vilken de bildar en bana för avsättningen av elastinet i de elastiska fibrerna. Även om allmänt förekommande i kroppen, finns mikrofibriller huvudsakligen i aorta, ledband och zonules av ciliära kroppar (vid okulär nivå).
Att vara en autosomal dominerande sjukdom påverkas bara barn som har ärft en FBN-1-gen som förändras av båda föräldrarna av Marfans syndrom. I ett fall av 4 är sjukdomen emellertid resultatet av spontana mutationer hos patienter som inte har släktforskning.
Sjukdomsnamnet kommer från den franska barnläkaren som först beskrev den 1896 (A. Marfan), varefter den var tvungen att vänta till 1991 för att identifiera den förändrade genen som var involverad i symptomatisk manifestation: upptäckaren var F. Ramirez.
Titta på videon
X Titta på videon på youtubeorsaker
Vi har nämnt att Marfan syndrom är det omedelbara uttrycket för mutationen av en gen som kodar för fibrillin-1. Låt oss försöka fördjupa ämnet för att förstå vilken mekanism som är etablerad för att utlösa syndromet.
FIBRILLINA 1 är en glykoproteinkomponent av elastin, väsentligt för att säkerställa och bibehålla elasticitet och vävnadsstyrka. I fysiologiska förhållanden binder fibrillin 1 till ett annat protein, känt som TGF-beta (eller transformerande tillväxt-beta-faktor). TGF-beta verkar vara involverad i skadliga processer som påverkar vaskulär glattmuskel och den extracellulära matrisen. Utgående från dessa antaganden är vissa författare övertygade om att Marfan syndromet förutom mutationen av FBN-1-genen också beror på ett överskott av TGF-beta, särskilt i aorta, i hjärtklaffarna och i lungorna.
incidens
Det beräknas att Marfan syndrom påverkar 1 patient varje 3000-5000 födda och manifesterar sig otydligt mellan män och kvinnor, utan predilektion för en exakt rasen. Statistiken visar att 75% av patienterna har en positiv familjehistoria; i de återstående 25% ligger orsaken till sporadiska mutationer som verkar vara associerade, på något sätt, med faders avancerade ålder vid tidpunkten för uppfattningen.
Barn med extremt svåra former av Marfan syndrom har en livslängd på mindre än ett år.
Före utvecklingen av öppenhjärtkirurgiska strategier hade de flesta patienter med Marfan syndrom en genomsnittlig livslängd på 32 år. Tack vare den ständiga förbättringen av medicinska och farmakologiska terapier lever Marfans syndrompatienter i genomsnitt upp till 60 år.
Tecken och symptom
Att lära sig mer: Symptom Marfan syndrom
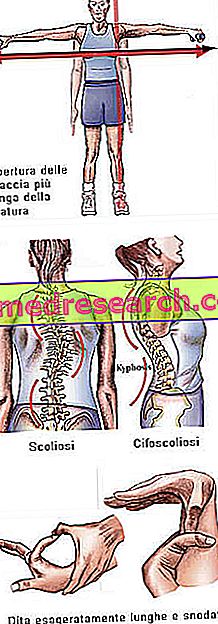
Marfan syndrom kan springa helt asymptomatisk. Intresserade patienter har en alltför slank struktur, som är oproportionerligt lång och tunn. De nedre och övre extremiteterna har en mycket högre längd än stammen (dolicostenomegalia). Det är också talat om att arachnodactyly bäst uttrycker begreppet överdriven längd av fingrarna, typiska för ämnen som påverkas av Marfan syndrom: händerna jämförs därför med benen på en spindel.
När det gäller höjd har dessa patienter en genomsnittlig höjd över 97: e percentilen.
Andra särdrag som ofta finns hos patienter med Marfan syndrom inkluderar:
- Vapen öppna större än höjd
- Lösa leder → överdriven gemensamma rörlighet
- Deformation av bröstväggen
- Kristallisk förskjutning
- Övre delen av kroppen mindre utvecklad än underdelen
- Spontan pneumotorax (11%)
- skolios
- Hudstriae på lår, rygg, deltoid, pectoral nivå
Bland de mest problematiska tecknen som är associerade med Marfan syndrom nämns hjärtfluktprolaps och mitralventilinsufficiens: ett liknande tillstånd kan enkelt främja utvidgning av aorta-ringen och aorta-dissektion.
Tabellen visar tecken som kan hittas hos patienter med Marfan syndrom. De tecken som beskrivs där finns inte alltid närvarande, men en bra del av dessa finns.
| Inverkan på anatomisk plats | Möjliga symptom |
söt | Striae i bröstkorg, ländrygg och sakrala område |
ögon | Nedsatt syn, astigmatism, retinal detachement, smalvinkelglaukom, linsförskjutning, myopi |
Benstruktur | Artralgi, kyphoskoliosi, dolicostenomelia (överdriven utveckling i benlängd i förhållande till stammen), hypermobilitet, högt gommen, deformerad bröstkorg, plana fötter, smala och tunna handleder, onormal återkomst / sternumprotes, skolios, krökta axlar, spondylolistes |
Dita | arachnodactyly |
lungor | Spontan pneumothorax, dyspné, idiopatisk lungsobstruktiv sjukdom |
Ansiktsförändringar | Ogival gom (missbildning av gommen), mandibular retrognathia (defekt i käkens utveckling), långsträckt ansikte |
hjärta | Angina pectoris, abdominal aorta aneurysm, hjärtarytmi, bröstkörtelvätska / brutning / dissektion, aorta insufficiens, mitralventil prolapse |
språk | Språk svårigheter |
diagnos
Med tanke på mer än 200 möjliga mutationer är användningen av genetiska markörer nästan omöjlig för diagnostiska ändamål.
Uppskattningen av Marfan syndrom är inte alltid så omedelbar, eftersom fenotypisk uttryck av mutationen inte alltid är uppenbart och enkelt att identifiera. Diagnosförseningen kan allvarligt äventyra patientens överlevnad: tänk bara på att du inte känner igen ett kardiovaskulärt problem.
Diagnostiska kriterierna för Marfan syndrom utarbetades internationellt 1996: diagnosen består av utredning av familjehistoria i samband med en kombination av större och mindre indikatorer på syndromet.
Några av de många diagnostiska testerna som används är:
- ekokardiogram
- Magnetisk angiorison och CT (för undersökning av aorta)
- magnetisk resonansangiografi (MRA) med kontrastvätska (för att framställa interna strukturer i aortan)
- undersökning med slitlampor (för att analysera eventuell dislokation av linsen)
- mätning av okulärt tryck (för att markera eventuell närvaro av glaukom)
- genetiska tester (rekommenderas innan du tar ett barn för att bestämma syndromet eller inte)
terapier
Att vara en genetisk sjukdom, det finns inget läkemedel eller en behandling som kan vända sjukdomen.
Användningen av droger är emellertid avgörande för att mildra symtomen och undvika komplikationer, i synnerhet hjärtliga. För detta ändamål är läkemedel för att minska blodtrycket, såsom sartaner (speciellt), ACE-hämmare och beta-blockerare, särskilt lämpliga.
I samband med Marfan syndrom kan patienter som lider av skolios också följa särskild vård, liksom för de som drabbats av glaukom.
Kirurgi är tänkbart att korrigera den anomala aorta dilatationen, ett element som ofta förenar majoriteten av patienterna med Marfan syndrom.
Fortsätt: Marfans syndrom - Läkemedel och vård »